
When Stephen Tower’s right hip gave out in 2006, he asked his surgeon to implant an artificial one — specifically, a metal-on-metal hip called the ASR XL, made by Johnson & Johnson. He knew what he was talking about: As an orthopedic surgeon, Dr. Tower specializes in complex hip replacements. But what he knew wasn’t enough to protect him from a defect in the device.
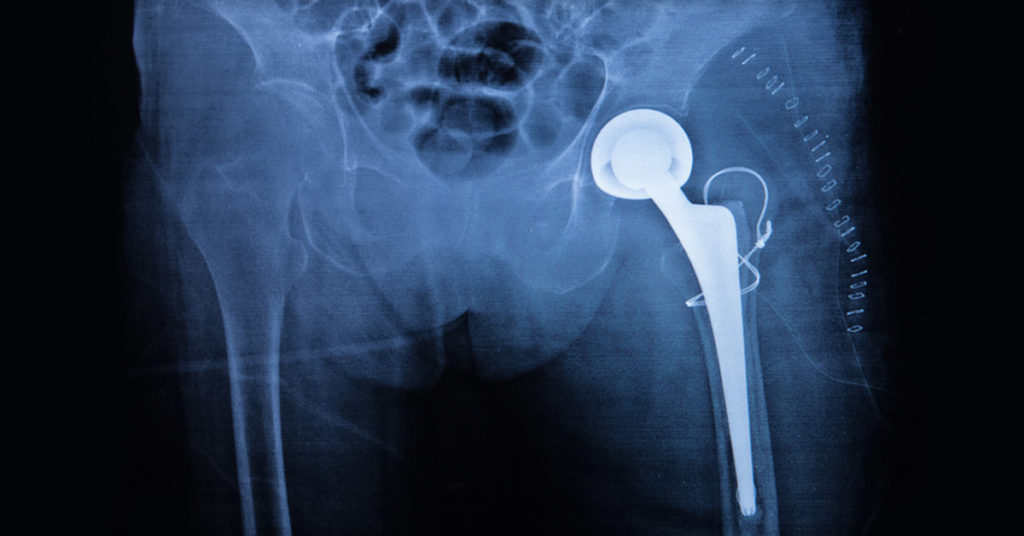
Five years after his surgery, and in excruciating pain, Dr. Tower underwent more surgery, this time to have the device replaced. When the surgeon sliced into his hip, what he saw looked like a crankcase full of dirty oil. Tissue surrounding the hip was black. Cobalt leaking from the ASR hip had caused a condition called metallosis, destroying not only local muscle, tendons and ligaments, but harming Dr. Tower’s heart and brain as well.
Metal-on-metal devices
Non-MOM Devices
Despite Dr. Tower’s repeated efforts to warn his colleagues and the company that the implants were harming patients, Johnson & Johnson continued to market metal-on-metal hips. While it withdrew the ASR XL model from the market in 2010, citing slow sales, it continued to sell another, similarly problematic model, the Pinnacle, until 2013.
More than 9,000 patients filed suit against the company, and on Nov. 16, six New York patients won a $247 million trial verdict for serious harms caused by the Pinnacle hip implants and for failing to warn doctors and patients about its dangers. These suits and others are pulling back the curtain on what some doctors call the Wild West of medicine: the untested and largely unregulated medical device industry.
About 32 million Americans — or about one in 10 — have at least one medical device implanted, from artificial joints to cardiac stents, surgical mesh, pacemakers, defibrillators, nerve stimulators, replacement lenses in eyes, heart valves and birth control devices.
These devices have helped countless people, and some have saved lives. But many others are harmed — and doctors and patients are at the mercy of manufacturers’ claims about the safety and efficacy of the devices. Medical interventions are now the third-leading cause of death in the United States, and devices play an increasing role in that statistic.
Many people assume that the Food and Drug Administration requires rigorous testing of medical devices before they are approved, the same as the lengthy approval process it requires for new drugs. In fact, most high-risk devices on the market, including implants, have undergone no clinical testing at all.
Although the standard for approval of a new drug usually calls for two randomized, controlled clinical trials, the standard for many medical devices is no standard at all. Since medical devices didn’t come under regulatory control by the F.D.A. until 1976, the agency simply grandfathered in all devices that were already on the market under a provision known as 510(k), which allows manufacturers to sell most new devices without requiring any clinical testing as long as the manufacturer says its product is “substantially equivalent” to an existing device.
In addition to the 510(k) pathway, medical device companies can avoid clinical testing for the highest risk devices through the supplement pathway by telling the F.D.A. they made a minor change to a previously approved device. The use of these loopholes is widespread: A study published in The Journal of the American Medical Association in 2009 found that only 5 percent of high-risk implanted cardiac devices even partly met the standard for drug testing.
Metal hips are far from the only devices with catastrophic consequences. In October 2007, Medtronic, a leading medical device manufacturer, recalled the lead wires in its Sprint Fidelis defibrillator after they were found to fracture and misfire, harming or even killing patients. The devices had not been clinically tested and were approved for sale by the F.D.A. through the supplement pathway. But in this case, the “minor change” was a fatal one; the new wire was thinner and prone to fracture.
By the time of the recall, 268,000 leads had been implanted in patients worldwide, the majority in the United States. After the recall, many patients rushed to have the devices removed, but removal posed its own dangers, causing major complications in 15 percent of patients.
Even when devices are subjected to trials, the F.D.A. sometimes ignores danger signs detected by those studies. In 1997, during the approval process of the vagus nerve stimulator, a device made by Cyberonics to treat epilepsy, an F.D.A. adviser voiced concerns about a high death rate noted in patients with the device. But the agency didn’t stop the device from going to market. Instead, it awarded conditional approval, meaning that Cyberonics would have to conduct safety studies after the device was on the market.
The agency didn’t even require Cyberonics to inform patients that there was concern about the death rate, or that they were effectively being made unwitting guinea pigs. When Cyberonics finally submitted five studies that it said proved the device was safe, it failed to include death data for any of the studies, a move the F.D.A. defended, saying the agency hadn’t asked the company to count deaths, only to “characterize” deaths.
How it’s possible to characterize deaths without including any actual data on deaths is anyone’s guess.
With such shockingly lax regulations, it’s no surprise that device recalls have risen over the years; in 2003, there were eight Class 1 device recalls, which the F.D.A. defines as indicating “a reasonable probability” that a device will “cause serious adverse health consequences or death.” In 2016, that number rose to 117, affecting hundreds of thousands of patients.
Loose oversight of devices poses a threat to public health. Yet the Trump administration’s picks to lead the Department of Health and Human Services and the F.D.A. are advocates of deregulation, which they insist will speed the march of “lifesaving” devices to market. This presumes that such speed and innovation is actually lifesaving or life-enhancing. The number of device-related deaths shows how dangerous that presumption is.
The fact that the F.D.A. has allowed lax practices like the 510(k) loophole to continue, despite calls for reform by institutions like the National Academy of Medicine, poses the question of why the F.D.A. would put profits ahead of safety. One clue rests in history: In 1988, President Ronald Reagan changed the position of F.D.A. commissioner from civil servant to political appointee, which has meant that presidents, with their war chests fattened by the drug and device industries, have repeatedly appointed industry-friendly commissioners, with rare exception.
Returning to a system in which F.D.A. commissioners are civil servants is perhaps a first step, but it needs to be the first of many. As a government agency, the F.D.A. is supposed to serve as a bulwark between corporate profiteering and the public welfare. Its continued allowance of regulation loopholes like 510(k), despite the significant demonstrated cost to public welfare, calls into serious question the agency’s fulfillment of its stated mission to protect public health by ensuring safety, efficacy and security.
Jeanne Lenzer is the author of “The Danger Within Us: America’s Untested, Unregulated Medical Device Industry and One Man’s Battle to Survive It.”
Originally Published in the New York Times